Selected Publications:
For a complete list of publications, please visit Pubmed.gov ![]()
|
|
A. Lorzadeh, C. Hammond, F. Wang, D. J. H. F. Knapp, J. CH. Wong, J. Y. A. Zhu, Q. Cao, A. Heravi-Moussavi, A. Carles, M. Wong, Z. Sharafian, J. Steif, M. Moksa, M. Bilenky, P. M. Lavoie, C. J. Eaves & M. Hirst. Polycomb contraction differentially regulates terminal human hematopoietic differentiation programs BMC Biology 2022 May 13. | 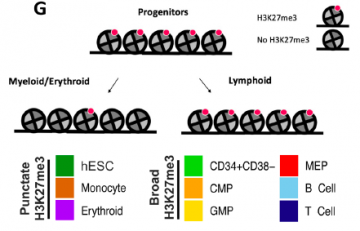 |
|
|
Lussier AA, Bodnar TS, Mingay M, Morin AM, Hirst M, Kobor MS, Weinberg J. Prenatal Alcohol Exposure: Profiling Developmental DNA Methylation Patterns in Central and Peripheral Tissues. Front Genet. 2018 Dec 4;9:610. doi: 10.3389/fgene.2018.00610. eCollection 2018. |  |
|
|
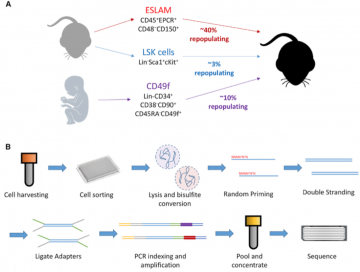
This Stem Cell Reports paper present an analytical strategy (PDclust) developed in Dr. Hirst lab to define single-cell DNA methylation states through pairwise comparisons of single-CpG methylation measurements:
Hui et al., High-Resolution Single-Cell DNA Methylation Measurements Reveal Epigenetically Distinct Hematopoietic Stem Cell Subpopulations, Stem Cell Reports (2018), https://doi.org/10.1016/j.stemcr.2018.07.003 |
 |
|
|
In this Nature paper, the authors applied a technology developed in Dr. Hirst lab to track individual cells to study clonal evolution in human glioblastoma. They report the clonal evolution of barcoded glioblastoma cells in an unbiased way following serial xenotransplantation to define their individual fate behaviours. | 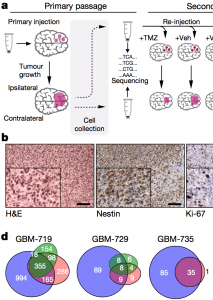 |
|
|
In this publication we reveal a specific role for vitamin C in histone demethylation in ES cells and document that DNA methylation and H3K9me2 cooperate to silence germline genes in pluripotent cells. | 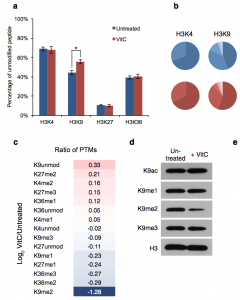 |
|
|
We report here the first use of whole-genome sequencing (WGS) to examine the initial clonal dynamics in an unusual patient with chronic myeloid leukemia (CML). Whole-genome analysis reveals unexpected dynamics of mutant subclone development in a patient with JAK2-V617F-positive chronic myeloid leukemia, Experimental Hematology 2017;53:48–58 |
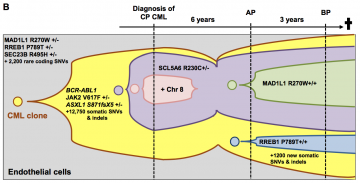 |
|
|
Matthew Mingay et al. provide a survey of the chromatin landscape of HOXA9-IDH1R132H AML and new findings supporting a role for vitamin C in facilitating the epigenetic remodelling that occurs during differentiation of hematopoietic progenitors (Vitamin C induced epigenomic remodelling in IDH1 mutant acute myeloid leukemia, Leukemia accepted article preview 2 June 2017; doi: 10.1038/ leu.2017.171.) |  |
|
|
As part of the International Human Epigenome Consortium (IHEC), our team investigated the relationship between bivalent histone modifications and nucleosome density by utilizing a native ChIP approach (Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility. Cell reports 17, 2112–2124 – November 15, 2016). In the same issue, we report an epigenomic comparison of three cell types of normal adult human mammary gland, with their associated stromal cells, and with three immortalized, non-tumorigenic human mammary cell lines. (Analysis of Normal Human Mammary Epigenomes Reveals Cell-Specific Active Enhancer States and Associated Transcription Factor Networks – Cell Reports 17, 2060–2074, November 15, 2016). |   |
|
|
Explore consortium data at the Cell Press IHEC webportal at http://www.cell.com/consortium/IHEC. The papers in this Cell issue reveal DNA and histone modifications, nucleosome positioning, and chromatin architecture in primary human cells. These findings provide critical new insight into cell-type-specific biology, variation between individuals, and disease (Hendrik G. Stunnenberg, The International Human Epigenome Consortium, and Martin Hirst – The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery -Cell 167, November 17, 2016)
This SnapShot depicts key sequencing-based methods used in the analysis of epigenomes, including (1)bisulfite sequencing, (2) chromatin immunoprecipiation sequencing, (3) determination of open chromatin, and (4) 3D chromatin capture (SnapShot: Epigenomic Assays- Martin Krzywinski, Martin Hirst – Cell, Vol. 167, Issue 5) |
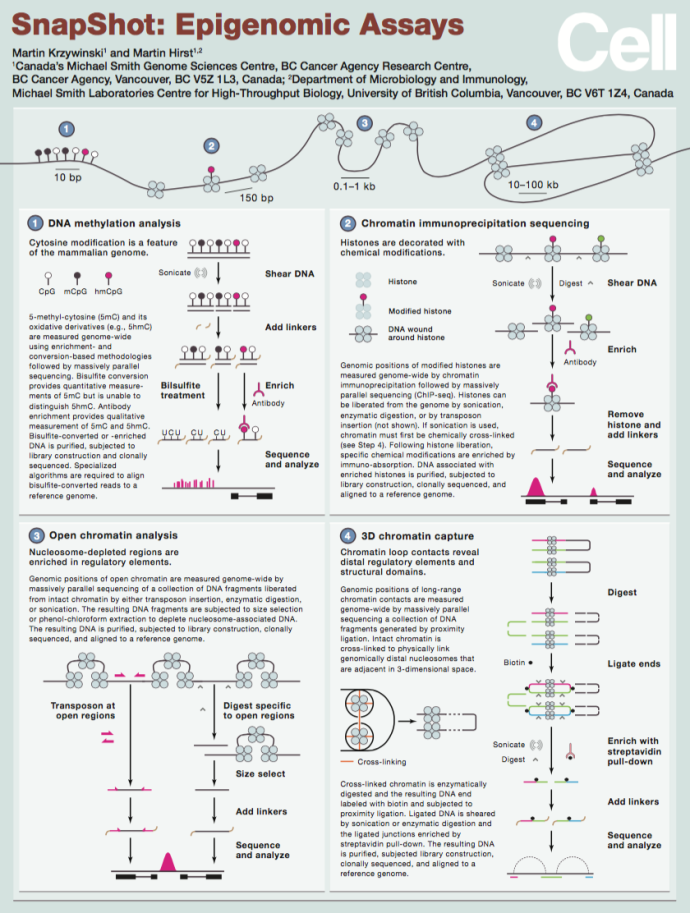 |
|
|
Alireza Lorzadeh et al. provide a robust approach for combining genome-wide methylation profiles of histones with MNase accessibility. Application of this technology to primitive human cord blood and embryonic stem cells indicates differences in histone modifications and their relationship to transcription between these two cell types (Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility. Cell reports 17, 2112–2124 – November 15, 2016) | 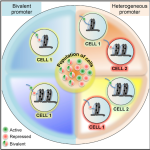 |
|
|
Davide Pellacani et al. present comprehensive histone and DNA modification profiles for four cell types in normal human breast tissue and three immortalized human mammary epithelial cell lines. Analysis of activated enhancers place luminal progenitors in between bipotent progenitor-containing basal cells and nonproliferative luminal cells (Analysis of Normal Human Mammary Epigenomes Reveals Cell-Specific Active Enhancer States and Associated Transcription Factor Networks – Cell Reports 17, 2060–2074, November 15, 2016) | 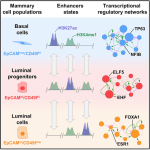 |
|
|
Our research involves comprehensive genome-wide analyses. In this study of the SMARCB1 loss in malignant rhabdoid tumors (MRTs), whole-genome sequencing, whole-genome bisulfite sequencing, whole transcriptome (RNA-seq), microRNA sequencing (miRNA-seq), and histone modification profiling were integrated to characterize extra-cranial MRTs. Our analyses revealed gene expression and methylation subgroups and focused on dysregulated pathways, including those involved in neural crest development (Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell. 2016) |  |
|
|
As part of the NIH Roadmap Epigenomics Consortium, our lab participated in the integrative analysis of 111 reference human epigenomes, profiled for histone modification patterns, DNA accessibility, DNA methylation and RNA expression. Global maps of regulatory elements were established, regulatory modules of coordinated activity, and their likely activators and repressors were defined. The results provide a resource for interpreting the molecular basis of human disease and demonstrate the central role of epigenomic information for understanding gene regulation, cellular differentiation and human disease (Roadmap Epigenomics Consortium et al. Nature 518, 317-330 (2015) doi:10.1038/nature14248) |  |
|
|
Based on epigenomic and transcriptional profiles generated from primary human breast cell types isolated from disease-free human subjects, our lab defined a regulatory network for luminal and myoepithelial cells and provided an intersection with breast cancer genome-wide association study alleles. A set of myoepithelial- and luminal epithelial-specific intronic retention events was also characterized. Our findings provide novel mechanistic clues about the events that contribute to normal epithelial differentiation and provide a comprehensive reference for future studies of disease (Epigenetic and transcriptional determinants of the human breast- Nature Communications Feb. 2015 doi:10.1038/ncomms7351) |  |
|
|
Our lab is involved in the construction of barcode amplicon libraries for massive parallel sequencing (MPS). We developped an analysis pipeline for computational processing of raw sequencing data from barcoded samples. The lentiviral genomic-barcoding methodology allows analyzing clones in regenerating epithelial populations (Clonal analysis via barcoding reveals diverse growth and differentiation of transplanted mouse and human mammary stem cells. Cell Stem Cell. 2014 Feb 6) or following human mammary cells transduced with an oncogene (Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature 2015) or tracking the number and size of multiple subclones within a single human tumour xenograft and their response to continued in vivo passaging (DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat Commun. 2014 Dec 23;5:5871. doi: 10.1038/ncomms6871) | 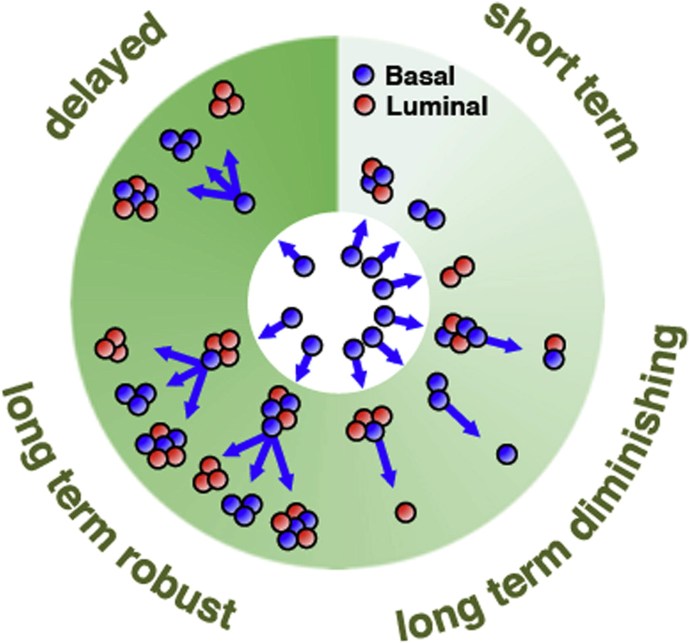 |